在全球医疗器械行业中,欧盟市场始终以严格、透明且系统化的法规体系著称。
随着《欧盟医疗器械法规》(Regulation (EU) 2017/745,简称MDR)自2021年正式全面实施,对于希望进入欧盟市场的企业而言,理解MDR的结构框架、监管主体、合规路径以及注册资料要求,已成为所有国际化布局的基础工作。为了帮助行业更好地理解MDR本身,本文将从监管机构、法规体系、产品分类以及注册流程等关键环节进行系统梳理。
欧盟医疗器械合规:多层级协同的监管体系
要理解MDR的执行逻辑,首先需要明确欧盟医疗器械监管系统的角色分工。欧盟整体采用“立法统一、执行分散、协调补强”的模式,而这也是MDR能够在27个成员国中保持统一性的关键。
监管中,欧盟委员会(European Commission)负责法规的制定、修订与解释,是整个MDR体系的核心推动者。

而在法规落地层面,则由各成员国的主管当局(Competent Authorities)承担市场监管、企业稽核、不良事件处理等职责,这些主管机关具有高度自治性,却又必须遵循欧盟层面的统一要求。
与此同时,位于企业合规核心环节的,是由各国指定并由欧盟认可的第三方机构——Notified Body(公告机构)。中高风险类医疗器械必须通过公告机构的符合性评定后才能贴上CE标识。
欧盟医疗器械法规:从指令迈向法规时代
在MDR实施之前,欧盟医疗器械合规基于若干指令(Directive),即MDD(93/42/EEC)、AIMDD(90/385/EEC)等。指令的特点是必须由各国转换成本国法律,因此在各成员国执行细节上可能存在差异。
MDR的出现彻底改变了这一点,MDR具备直接效力,无需各国立法转译即可直接实施。这使得欧盟医疗器械监管在统一性、可预测性以及执行力上大幅提升。

除了MDR本身,体外诊断医疗器械还受到独立法规IVDR(Regulation (EU) 2017/746)的管理,与MDR并行构成欧盟医疗器械监管体系的两大支柱。在MDR的框架之下,产品分类、临床评估、经济运营者责任、市场监督与可追溯性都被系统化纳入统一要求。
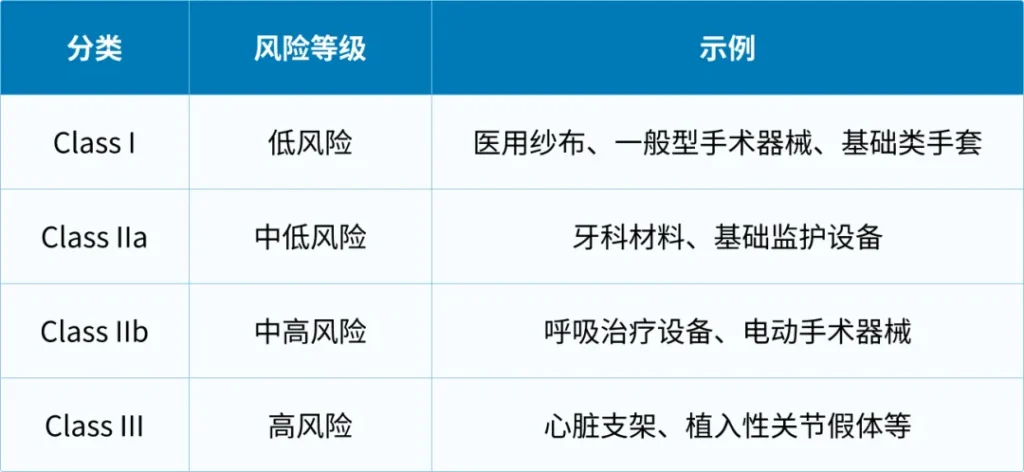
欧盟医疗器械产品分类:基于风险原则的四级体系
欧盟采用基于风险的医疗器械分类体系,将器械分为Class I、Class IIa、Class IIb、Class III四级。
不同类别器械评定差异:
- Class I多为低风险产品,部分可由企业自我声明合规;
- Class IIa/IIb属于中高风险,则必须通过公告机构的审核;
- Class III为最高风险等级的植入类或生命维持类设备,需要完整的临床证据和最严格的评定流程。
分类依据产品的预期用途、侵入性、与人体接触方式、能量来源以及风险属性,具体规则详见MDR附件VIII。
欧盟医疗器械合规:注册流程及资料清单
在MDR体系中,医疗器械进入欧盟市场并非单一步骤,而是一个覆盖企业信息、产品注册、临床数据、符合性评定以及上市后监管的完整流程。
整体而言,可分为以下几个阶段:
首先,制造商、进口商或授权代表必须在EUDAMED中完成Actor Registration(角色登记),并获得统一的SRN(Single Registration Number)。这一步是企业进入欧盟体系的基础身份认证,也是后续所有注册步骤的前提。
其次,企业需按照MDR附件VII完成产品分类,并以此确定是否需要公告机构参与的CE合格评定流程。对于中高风险器械(IIa/IIb/III),公告机构将审核企业的质量管理体系、技术文档以及临床评估报告。在此阶段,技术文档是审核核心,其中包括风险管理、设计控制、制造流程、产品规格、标签信息、UDI数据等内容。
当器械通过符合性评定后,制造商需在EUDAMED完成UDI/Device Registration(器械注册),包括UDI-DI、Basic UDI-DI、型号与产品特征信息等。之后,企业方可正式在欧盟市场投放产品。
然而,合规并不随着上市而结束。MDR强调上市后监管(PMS)和警戒系统(Vigilance),企业需持续提交安全更新报告(PSUR)、分析市场风险、监测不良事件,并在必要时采取纠正措施。欧盟通过EUDAMED对这些信息进行集中管理,使整个市场透明有效。
MDR注册文件大致包括:
- 企业信息(制造商/授权代表/进口商资料)
- 质量管理体系文件(通常为ISO 13485基础)
- 风险管理报告(符合ISO 14971)
- 临床评估报告(CER)
- 技术文档(包括设计、制造、验证、说明书、标签等)
- UDI数据
- 上市后监管计划和持续更新资料(如PMS、PSUR)
欧盟医疗器械CE认证
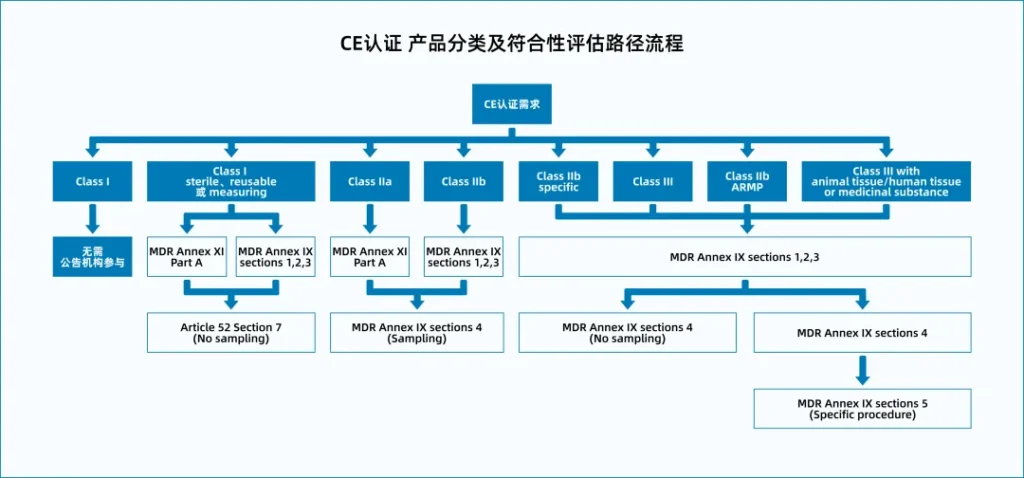
在完成注册流程与技术文档准备之后,医疗器械真正迈向欧盟市场的关键一步,就是取得 CE认证。CE标志意味着产品已经满足欧盟《医疗器械法规》(MDR 2017/745)的安全与性能要求,可以在欧盟及欧洲经济区自由销售。需要注意的是,CE并不是单一证书,而是一套风险管理、临床评价、质量体系与上市后监测共同构成的合规体系。

由于欧盟采用以风险等级为基础的管理逻辑,因此不同类别的医疗器械,其CE路径也不同:
Class I多数可通过自我符合声明完成,而Class IIa、IIb、III则必须由公告机构(Notified Body)参与审核。
这些差异直接决定了所需提交的技术文档深度、审核方式与认证周期,也因此成为许多企业制定合规策略时的关键考量因素。
对于企业而言,CE标志不仅是进入欧盟市场的通行证,更是产品已经通过严格法规体系验证的重要证明。拥有CE的医疗器械,可以在欧盟内部自由流通,也更容易获得其他认可CE的国家和地区的市场准入。同时,CE认证要求建立完整的质量管理体系、风险管理体系和上市后监管机制,使企业在产品全生命周期管理上更具系统性与可追溯性。
MDR的出现不仅是监管升级,更代表着欧盟对医疗器械行业“安全优先、透明为本、全过程监管”的坚定态度。对于企业而言,虽然合规门槛提高,但市场的规范化与透明化也为真正有实力、有质量管理基础的企业带来更公平且可持续的竞争环境。
恩特服务内容
加拿大、美国等地区医疗器械合规服务可与我们联系