随着全球医疗技术的快速迭代,越来越多企业希望进入美国这一重要且合规复杂的医疗器械市场。
然而,FDA的监管体系并不是简单的注册流程,而是一套覆盖产品设计、技术审查、质量体系到上市后监管的全生命周期管理机制。
对于计划进入美国市场的企业来说,理解这一体系,不只是为了“过审”,更是为了建立长期竞争力。
美国医疗器械 | 从法律到监管机构
美国的医疗器械监管体系由 FDA 负责,具体由其下属的CDRH(Center for Devices and Radiological Health)执行。整个监管体系的核心依据来自《食品药品和化妆品法》(FD&C Act),并辅以大量细化的法规条款。
值得注意的是,FDA 目前正推动质量体系从传统的21 CFR Part 820(QSR)过渡到QMSR,使其与ISO 13485更紧密衔接。这意味着未来的美国医疗器械质量要求将更“国际化”。
美国医疗器械 | 产品分类决定企业合规之路
美国对医疗器械采取“风险分类”制度,这决定了后续所有的审查深度与资料要求。以下是一张简洁的对照表,帮助你快速理解分类差异:
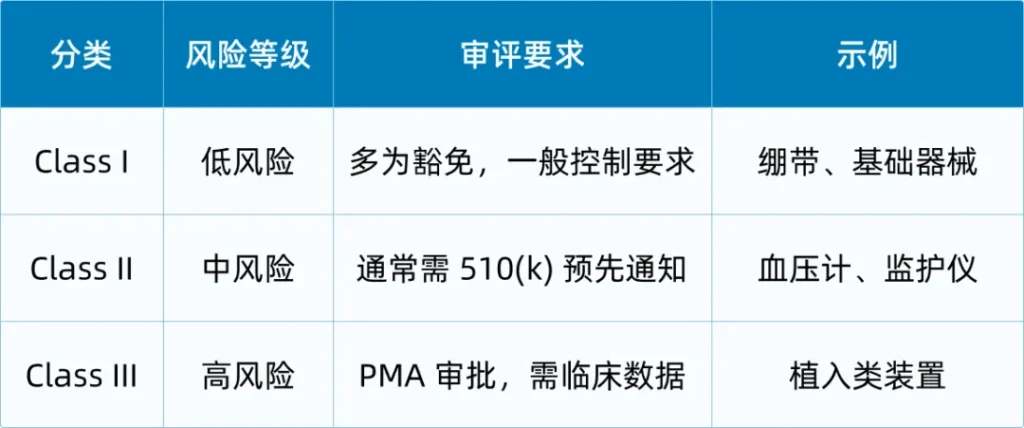
对于一些市场上没有“等同器械”可参照,但风险并不高的创新产品,企业也可以选择De Novo路径,这在过去几年已成为创新器械常用的合规方式。
美国医疗器械 | 三重审查 510(k)、PMA与De Novo
上市前通报 510(k):在美国医疗器械准入体系中,最受关注的莫过于 510(k)。如果产品能找到一款已合法上市的器械作为对照(即“等同器械”),并证明两者在安全性和有效性上“实质等效”,即可通过这一路径进入市场。
上市前批准 PMA(Premarket Approval):但对于那些风险更高或属于突破性创新的产品,FDA的审批则要求更严格。PMA不仅关注研究数据,更关注制造流程、风险管理和临床证据,是一道更高的准入门槛。
De Novo:当产品未能找到合适的等同器械,但风险又未达到Class III级别时,De Novo便成为一个“中间途径”,既兼具创新性,也能避免过度监管,因此常用于新型中低风险器械。
美国医疗器械 | 合规流程及资料清单
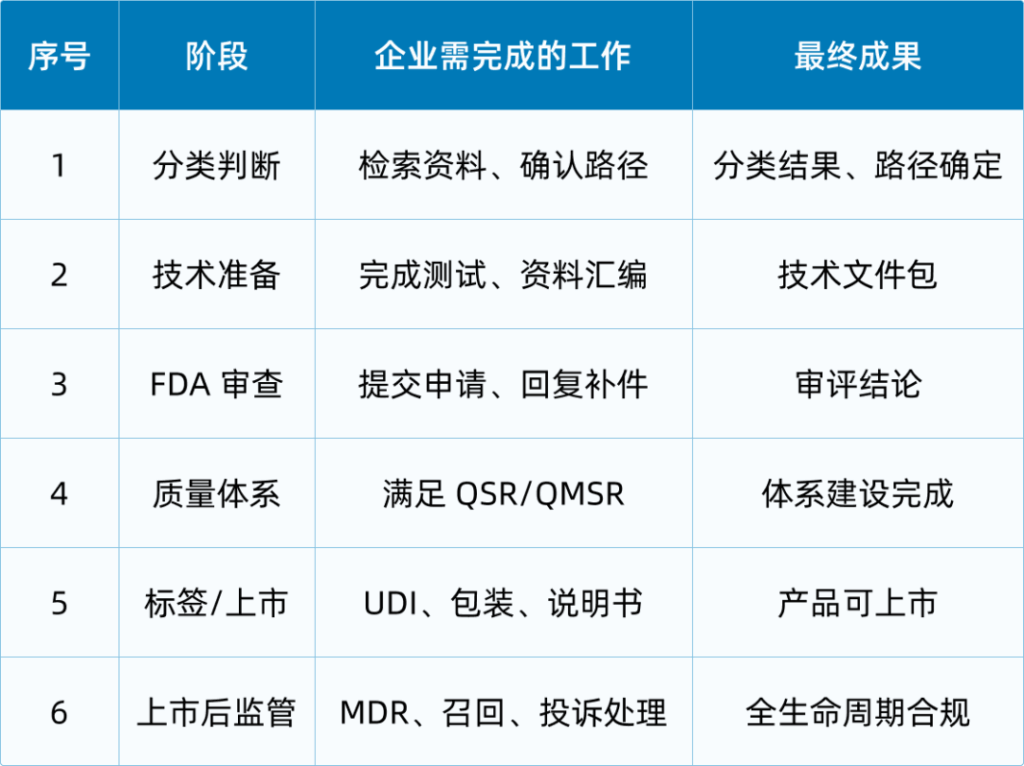
真正进入流程后,企业会发现,美国医疗器械的合规体系是一条覆盖产品全生命周期的路线,从设计、测试、临床,到审查、注册,再到上市后的追踪、投诉与召回管理,几乎没有“空白区”。
流程大致包括以下阶段:
确定分类与监管路径
- 确定分类与监管路径:查找数据库、确认是否有等同器械、判断是否需要510(k)、PMA或De Novo。
- 准备技术资料:包含设计、材料、性能测试、生物相容性、电气安全、风险管理、标签等核心内容。
- 进入FDA审查阶段:根据路径不同,可能会收到补充资料要求,企业需及时回复。
- 完成企业注册与产品列名:根据路径不同,可能会收到补充资料要求,企业需及时回复。
- 上市后监管:包括MDR事件报告、召回机制、投诉管理、质量体系维护等,是企业长期经营的关键环节。
510(k)提交资料清单(Premarket Notification)
根据FDA的要求,进行510(k) 提交时,通常需要准备以下资料:

很多企业在讨论“进入美国市场”时,更关注的是“拿证”,但真正构成壁垒的往往是质量体系能力、风险管理能力、持续合规能力。
对医疗器械行业而言,合规不是成本,而是品牌、信誉、技术实力的体现。
当体系真正建立起来,企业在国际市场的竞争力也会随之显著提升。
恩特服务内容
美国医疗器械注册申请/生产质量体系:
❑ 美国授权代表服务
❑ 生产企业FDA首年工厂注册/年度工厂注册
❑ 生产企业邓白氏码申请
❑ 美国市场产品上市申请/510(k)
❑ 美国市场产品分类界定/513(g)
❑ 美国市场产品创新产品上市申请/De Novo
❑ 符合美国FDA要求的医疗器械工厂生产质量体系建设/QSR820
❑ 产品注册前与FDA官方咨询沟通服务
❑ 符合美国FDA要求的医疗器械唯一标识(UDI)执行