主要法规
《医疗器械监督管理条例》
《医疗器械注册与备案管理办法》
《医疗器械产品技术要求编写指导原则》
《医疗器械说明书和标签管理规定》
《重组胶原蛋白创面敷料注册审查指导原则》
《医用透明质酸钠创面敷料注册审查指导原则》
《医疗器械申报资料要求和批准证明文件格式》
《医疗器械临床评价技术指导原则》
医疗器械定义
医疗器械,是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件;其效用主要通过物理等方式获得,不是通过药理学、免疫学或者代谢的方式获得,或者虽然有这些方式参与但是只起辅助作用;其目的是:
(一)疾病的诊断、预防、监护、治疗或者缓解;
(二)损伤的诊断、监护、治疗、缓解或者功能补偿;
(三)生理结构或者生理过程的检验、替代、调节或者支持;
(四)生命的支持或者维持;
(五)妊娠控制;
(六)通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息。
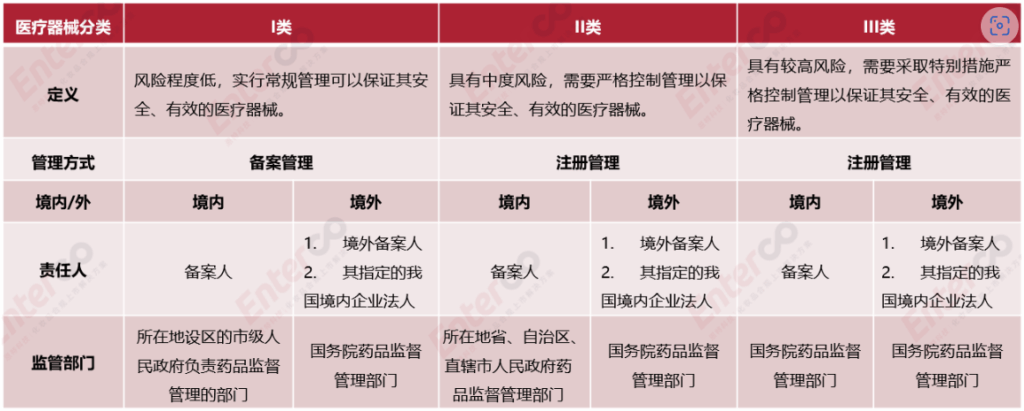
根据《医疗器械监督管理条例》,第一类医疗器械实行产品备案管理,第二类、第三类医疗器械实行产品注册管理。在投放中国市场前,应先向相应药品监督管理部门提交备案或注册申请并获得医疗器械备案凭证或注册证。同时确保在医疗器械生产质量管理规范体系下开展研发及生产经营活动。
中国医疗器械分类与监管
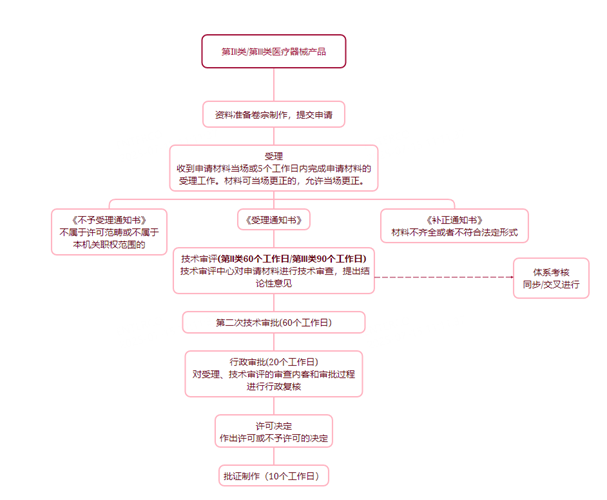
第II / III类医疗器械 – 产品注册流程
第II / III类医疗器械 – 产品注册资料要求
- 产品风险分析资料;
- 产品技术要求;
- 产品检验报告;
- 临床评价资料;
- 产品说明书以及标签样稿;
- 与产品研制、生产有关的质量管理体系文件;
- 证明产品安全、有效所需的其他资料。
服务内容
- 第一类医疗器械备案
- 第二类医疗器械注册
- 第三类医疗器械注册
- 进口医疗器械注册
- 体外诊断试剂注册
- 医疗器械延续注册
- 医疗器械变更注册及备案
- 医疗器械注册体系考核
- 医疗器械生产许可证
- 医疗器械法规培训